Dozens of FDA COVID-19-Related Guidances Will Soon No Longer Be in Effect. Are You Ready?

M. Jason Brooke, Esq.*
May 3, 2023
The FDA has issued over 80 COVID-19-related guidances since 2020, when the former HHS Secretary declared a public health emergency (PHE) related to COVID-19 under section 319 of the Public Health Service Act. Twenty-two of these guidances will no longer be in effect 180 days after the COVID-19 PHE declaration expires. The HHS recently announced its plan for the COVID-19 PHE declaration to expire at the end of the day on May 11, 2023, meaning these COVID-19-related guidances will no longer be in effect after November 7, 2023. The FDA issued final guidance, Transition Plan for Medical Devices That Fall Within Enforcement Policies Issued During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency (the “Enforcement Transition Guidance”), describing the FDA’s thinking on transitioning from the COVID-19-related enforcement policies described in these 22 guidances back to “normal operations.” The FDA’s transition process includes three phases, with the first and second phases occurring during the 180-day transition period, i.e., the period from May 11, 2023, when the COVID-19 PHE declaration is set to expire and Phase 1 begins, to November 7, 2023, when Phase 3 begins. During Phase 3, the FDA does not intend to object to the continued distribution of affected devices if the manufacturer has a marketing submission under review by the FDA.
Below I’ve tried to break down the salient information from the Enforcement Transition Guidance to help you sort out what the guidance says and how it might apply to you.
Does the COVID-19 PHE declaration expiration impact all of the COVID-19-related guidances?
No. The FDA identified 22 COVID-19-related guidances that will remain in effect for 180 days after the COVID-19 PHE declaration expires, meaning these 22 guidances and the COVID-19-related enforcement policies described therein will no longer be in effect after November 7, 2023. The FDA determined that stakeholders will need time to transition from the policies described in these affected guidances to normal operations, which the three-phase transition process outlined in the Enforcement Transition Guidance purports to provide.
The FDA identified 50 other COVID-19-related guidances that are not relevant to the three-phase transition process outlined in the Enforcement Transition Guidance:
- 22 guidances will no longer be in effect when the COVID-19 PHE declaration expires (i.e., May 11, 2023) because the recommendations are contained in other guidances or because stakeholders are no longer relying on the guidances;
- 24 guidances will remain in effect after the COVID-19 PHE declaration expires but will be superseded by a revised version, which the FDA intends to issue during the 180-day transition period; and
- 4 guidances that are not tied to the COVID-19 PHE declaration will remain in effect.
What are the COVID-19-related guidances that will remain in effect during the transition period?
The COVID-19-related guidances that will remain in effect during the transition period cover a variety of medical devices and digital health products, including non-invasive fetal and maternal monitoring devices, digital thermometers, digital therapeutics for psychiatric disorders, remote ophthalmic assessment devices, and digital pathology devices (among others).
What are the COVID-19-related guidances that will be revised during the transition period?
Of particular relevance, the FDA intends to revise the COVID-19-related guidances on policies related to clinical electronic thermometers, non-invasive remote patient monitoring devices, and face masks and face barrier coverings (not to be confused with face shields, surgical masks, and respirators). The rest of these guidances primarily relate to non-device policies.
When do the transition phases begin?
Manufacturers of medical devices that fall under the these soon-to-expire COVID-19-related enforcement policies should be aware of three approaching dates:
- May 11, 2023, the implementation date for FDA’s three-phase transition process and the start of Phase 1;
- August 9, 2023, the start of Phase 2 and the date by which manufacturers who intend to continue distributing their devices need to register and list; and
- November 7, 2023, the start of Phase 3 and the date by which manufacturers who intend to continue distributing their devices should have a marketing submission accepted by FDA.
More information on each of three phases is provided below.
What obligations for manufacturers occur during Phase 1?
During Phase 1, if not already doing so, manufacturers should follow 21 C.F.R. Part 803 (i.e., adverse event reporting requirements). Manufacturers who intend to continue distributing their devices after November 7, 2023 should begin preparing their marketing submissions. The FDA acknowledges certain “extenuating circumstances” that may exist, making it difficult for some manufacturers to submit an administratively complete marketing submission during the 180-day transition period (e.g., because of an ongoing clinical trial or longer term non-clinical study). The FDA encourages these manufacturers to engage the FDA through the Q-Submission Program early in the transition period. The FDA also acknowledges that there may be “unique compliance considerations,” particularly regarding Quality System (QS) requirements, that may warrant seeking an exemption or variance from a device QS requirement in accordance with 21 C.F.R. § 820.1(e). Manufacturers who intend to request an exemption or variance should submit their request within 90 days of the issuance of the Enforcement Transition Guidance, or by June 25, 2023.
Additionally, the FDA recommends that manufacturers of certain life-supporting or life-sustaining devices (identified in the Enforcement Transition Guidance and in Table 1 below) submit to the CDRH a “Notification of Intent” as soon as possible during Phase 1 (i.e., before August 9, 2023). The submission needs to contain the certain information about the manufacturer, the device, and a statement of whether the manufacturer plans to submit a marketing submission. If not planning to submit a marketing submission, the manufacturer is expected to discuss, as applicable, its plans to discontinue distribution of the device, to restore the device to an FDA-cleared or -approved version, to provide a physical copy and/or electronic copy of updated labeling, and any other efforts to address or mitigate potential risks of devices that remain distributed after Phase 2.
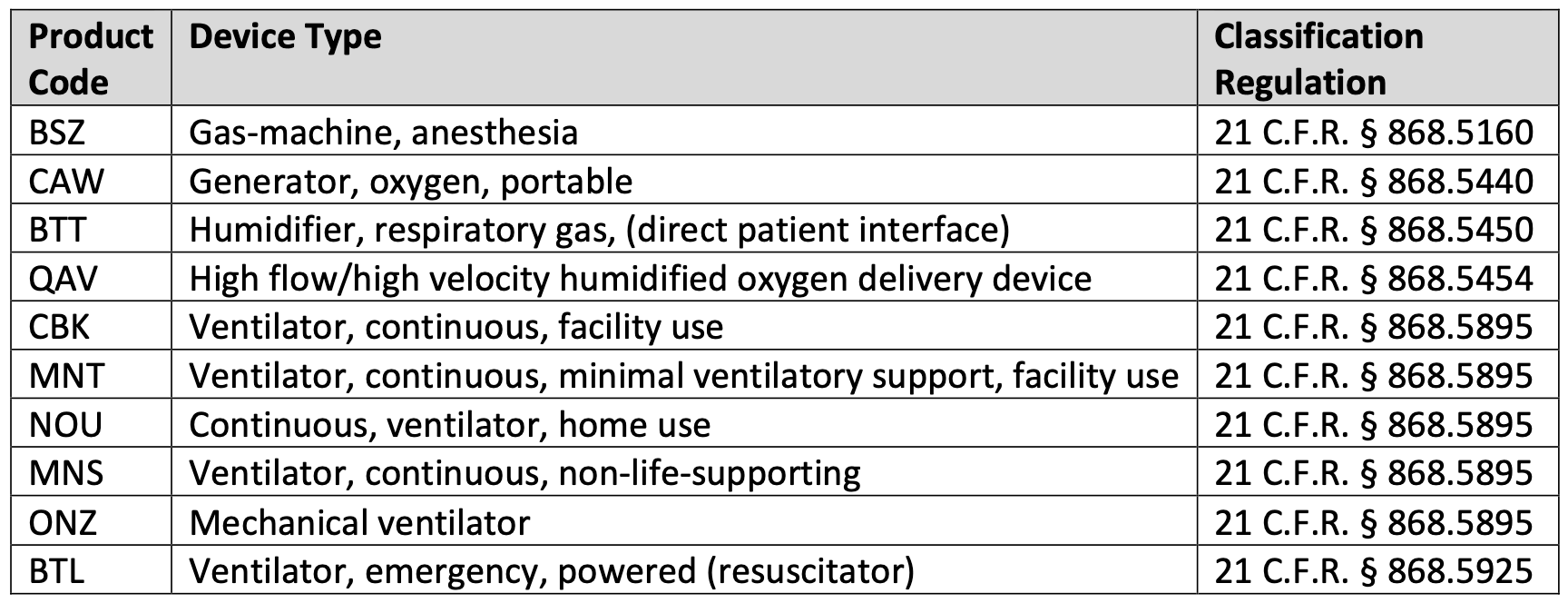
Table 1: Life-supporting or life-sustaining devices that require a Notification of Intent submission to the FDA.
What obligations for manufacturers occur during Phase 2?
During Phase 2, if not already doing so, manufacturers should follow 21 C.F.R. Part 806 (i.e., reports of corrections and removals requirements) and Part 807 Subparts B to D (i.e., registration and listing requirements).
Manufacturers who intend to continue distributing their devices after Phase 2 should complete and submit any required premarket submissions at least 3 weeks before the end of Phase 2 (due to the need for the FDA to review and accept the submission by November 7, 2023).
Manufacturers who do not intend to continue distributing their devices after Phase 2 should stop distribution before the end of Phase 2. The Enforcement Transition Guidance identifies three general circumstances in which the FDA does not intend to request market removal of already distributed devices:
- single-use, non-life-supporting/non-life-sustaining devices (e.g., face masks) that are used by the end user before the product expiration date;
- reusable, non-life-supporting/non-life-sustaining devices (e.g., infusion pumps) that are used by their end user and either are restored by the manufacturer to an FDA-cleared or FDA-approved version of the device or have a physical and/or electronic copy of updated labeling that accurately describes the product features and regulatory status (e.g., that the product lacks FDA clearance, approval, or authorization); and
- reusable life-supporting/life-sustaining devices (e.g., ventilators, extracorporeal membrane oxygenation systems) that are restored by the manufacturer to an FDA-cleared or FDA-approved version of the device so that they may be used by their end user; if not restored, the manufacturer should provide a physical and/or electronic copy of updated labeling that accurately describes the product features and regulatory status (e.g., that the product lacks FDA clearance, approval, or authorization) and such devices are not to be used.
What obligations for manufacturers occur during Phase 3?
After November 7, 2023, when the affected guidances are no longer in effect, the FDA does not intend to object to continued distribution of a device within the scope of the Enforcement Transition Guidance if a marketing submission for the device (or a device that is a derivative or the next generation of the device) is under review by the FDA, meaning that the premarket submission must have been sent to and accepted by FDA before the start of Phase 3 and the Agency has not taken a final action. Here, the term final action means a positive, negative decision, or notice of withdrawals related to the 510(k), De Novo, or PMA process.
For devices under review by the FDA during Phase 3, the FDA also does not intend to object to the device not complying with certain Unique Device Identification (UDI) requirements under 21 C.F.R. Part 801 Subpart B or other applicable labeling requirements under 21 C.F.R. Part 801 if the device continues to be labeled consistent with the relevant affected guidance. However, the FDA’s enforcement policy for these devices during Phase 3 does not extend to other requirements that may apply, such as registration and listing, QS, and reports of corrections and removals under 21 C.F.R. Parts 807, 820, and 806, respectively.
After the FDA takes a final action on the marketing submission (if it is not a negative decision), the FDA expects manufacturers to comply with all applicable regulatory requirements, including making updates to labeling and registration and listing information and complying with UDI requirements.
Are there “transition-specific” recommendations for marketing submissions?
Yes. The Enforcement Transition Guidance recommends that manufacturers include in the cover letters to their marketing submissions a statement that the device is/was distributed as described in the relevant COVID-19-related guidance, as well as submission number(s) for related premarket submissions. For manufacturers that register and list by the start of Phase 2, the FDA recommends using the term “enforcement” (as a shorthand for “enforcement policy”) in the premarket submission field if a submission number is not yet available.
The Enforcement Transition Guidance also recommends that manufacturers include in their marketing submissions a “Transition Implementation Plan,” describing the manufacturer’s plan for devices that are already distributed in the case of a positive decision or negative decision on the marketing submission. The Transition Implementation Plan should include, among other things:
- The estimated number of devices that are currently in U.S. distribution that fall within the scope of the affected guidances; and
- The process for notifying patients, consumers, healthcare facilities, healthcare providers, and device distributors of the device’s regulatory status.
In the event of a negative decision, if the manufacturer is proposing to leave already distributed product in place, the Transition Implementation Plan should address the rationale for doing so and considerations such as the following, where relevant:
- The process and timeline for restoring already distributed devices to an FDA-cleared or FDA-approved version;
- The process and timeline for providing a physical and/or electronic copy of updated labeling that accurately describes the product features and regulatory status (e.g., that the product lacks FDA clearance, approval, or authorization) for reusable devices.
- For reusable life-supporting/life-sustaining devices, the process for stakeholders to request and obtain a physical copy of updated labeling without additional cost; and
- A description of the maintenance plan for already distributed devices.
In the event of a positive decision, the Transition Implementation Plan should describe, as appropriate, the process and timeline for providing to users of already distributed devices updated labeling or components for the cleared or approved device, including updated labeling or components to reflect any cleared/approved changes to the already distributed device.
When should manufacturers stop distributing devices that fall within the 22 affected COVID-19-related guidances?
The Enforcement Transition Guidance states that devices that are “in distribution” are considered “already distributed” and defines “in distribution” to mean “finished, labeled devices that are no longer in the manufacturer’s possession that are in transit to or held in a third party’s device inventory not on behalf of the manufacturer, in a federal, state, or other government stockpile, or at a location where devices are then offered for direct sale to the end user.”
Manufacturers of devices distributed under the enforcement policies described in the 22 affected guidances who do not plan to continue distributing their devices or who fail to prepare and submit a marketing submission by the start of Phase 3 are expected to stop distribution by November 7, 2023. These manufacturers should be aware of any continuing legal requirements, such as adverse event reporting under 21 C.F.R. Part 803.
Manufacturers who prepare and submit marketing submissions before the start of Phase 3 and receive a negative decision from the FDA are expected to stop distribution when they receive the negative decision. Similarly, manufacturers who prepare and submit marketing submissions before the start of Phase 3 and either withdraw their submissions or fail to respond to the FDA’s request for additional information within the specified timeframe are expected to stop distribution by the date of withdrawal.
How does the expiration of the COVID-19 PHE declaration impact EUA-authorized devices?
Although this article focuses on the Enforcement Transition Guidance, it should be noted that the FDA issued a companion guidance titled, Transition Plan for Medical Devices Issued Emergency Use Authorizations (EUAs) Related to Coronavirus Disease 2019 (COVID-19) (EUA Transition Guidance). It’s important to note that the anticipated expiration of the PHE declaration on May 11, 2023 does not terminate an EUA declaration. An EUA, authorized under section 564 of the Federal Food, Drug, and Cosmetic Act (FD&C Act), remains in effect until the relevant EUA declaration terminates (or FDA revokes the specific EUA). In other words, termination of an EUA declaration terminates all EUAs issued under that declaration.
The FD&C Act requires advanced notice published in the Federal Register that an EUA declaration will terminate. The FDA does not plan to follow a three-phase transition process for devices authorized under a COVD-19-related EUA declaration. Rather, the EUA Transition Guidance states that the HHS intends to publish in the Federal Register advanced notice of each COVID-19-related EUA declaration termination on a date that is 180 days before the EUA-declaration termination date. During that 180-day period, manufacturers of EUA-authorized devices must continue to comply with the terms of the devices’ respective EUAs. Beginning on the relevant EUA-declaration termination date, EUA-authorized devices are no longer authorized for emergency use. The EUA Transition Guidance thus encourages manufacturers of EUA-authorized devices to prepare their post-EUA regulatory and disposition strategies now.
